Next: Desenvolvimento Up: Materiais e métodos Previous: Medidas estatísticas
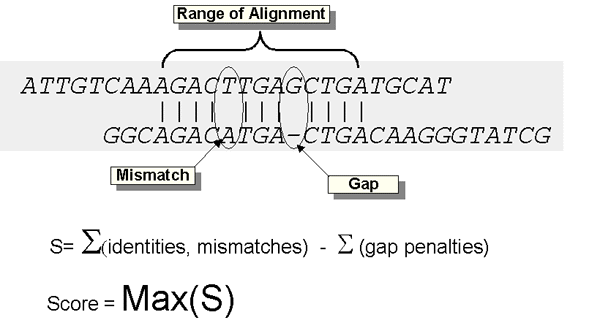
O Blast (Basic Local Alignment Search Tools) [2], desenvolvido pelo NCBI (National Center for Biotechnology Information), é um conjunto de algoritmos de comparação de seqüências de nucleotídeos e amino-ácidos. Segmentos de uma seqüência são comparados com segmentos de outras seqüências e são atribuídos pontos (scores) que indicam o grau de similaridade entre os segmentos. Quanto mais alta é a pontuação, maior é o grau de similaridade. Assim, ao final de uma execução do Blast temos, para cada par de seqüências, zero ou mais scores, dependendo do quão semelhantes elas são.
A seguir serão descritos alguns termos relevantes ao estudo do Blast:
Os valores desses custos são calculados empiricamente; em geral atribui-se um valor alto para a (10-15) e um baixo para b (1-2).
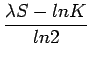
Onde ![]() e K dependem do sistema de pontuação (matriz de
substituição e tamanho das seqüências).
e K dependem do sistema de pontuação (matriz de
substituição e tamanho das seqüências).
Por ser uma versão normalizada do raw score, o bit score pode ser utilizado em comparações com scores de outros alinhamentos.
Ricardo Nishikido Pereira 2004-12-06